
- ABSTRACT:
- 1. INTRODUCTION
- 2. MATERIALS AND METHODS
- 2.1. Study Design
- 2.2. Target Protein Selection
- 2.3. Sequence Retrieval
- 2.4. Physicochemical Properties and Three-Dimensional Structure
- 2.5. Structural and Functional Domain Analysis
- 2.6. Active Site Identification
- 2.7. Ligand Selection and Preparation
- 2.8. Drug-Likeness Screening and ADME/T Prediction
- 2.9. Molecular Docking
- 2.10. Protein–Ligand Interaction Profiling
- 2.11. Lead Compound Identification
- 2.12. Reference Antibacterial Drug Selection
- 2.13. Molecular Dynamics Simulations
- 3. RESULTS
- 3.1. FASTA Sequence Retrieval
- 3.2. Physicochemical Characterization of Target Protein
- 3.3. 3D Structure Prediction of Target Protein
- 3.4. Functional Domain Identification of Target Protein
- 3.5. Active Site Identification
- 3.6. Ligand Selection
- 3.7. Virtual Screening of Ligands
- 3.8. Ligands ADME/T Properties
- 3.9. Molecular Docking
- 3.10. Interaction of Ligands and Target Protein
- 3.11. Lead Compound Identification
- 3.12. Comparative Investigation of Lead Compound vs. Cefotaxime
- 3.13. Absorption, Distribution, Metabolism, Excretion, and Toxicity Properties Comparison
- 3.14. Comparison of the Lipinski Rule of 5
- 3.15. Docking Score Comparison
- 3.16. Molecular Dynamics Simulations
- 4. DISCUSSION
- CONCLUSION
- AUTHORS' CONTRIBUTIONS
- ETHICAL APPROVAL & INFORMED CONSENT
- AVAILABILITY OF DATA AND MATERIALS
- FUNDING
- CONFLICT OF INTEREST
- ACKNOWLEDGEMENTS
- DECLARATION OF AI
- REFERENCES
Article ID: PD2601205002
Identification of Bioactive Postbiotics Against Neonatal Meningitis Caused by Group B Streptococcus via Srr2-Targeted In silico Screening
⬇ Downloads: 3
1Faculty of Health and Life Sciences, Capital University of Science and Technology, Islamabad, Pakistan
Received: 25 September, 2025
Accepted: 23 December, 2025
Revised: 08 December, 2025
Published: 06 May, 2026
ABSTRACT:
Background: Neonatal meningitis caused by Group B Streptococcus (GBS) remains a major contributor to morbidity and mortality, and rising antibiotic resistance motivates alternatives that spare the neonatal microbiome.
Method: Eight postbiotic metabolites (citric, lauric, oleic, linoleic, palmitic, palmitoleic, arachidonic, and malic acids) were screened against the GBS virulence factor Srr2 using blind docking (CB-Dock2/AutoDock Vina), pharmacokinetic/toxicity prediction (pkCSM), and 100-ns molecular dynamics (AMBER v22). The research compared the lead metabolite with cefotaxime.
Results: Citric acid showed the best docking affinity to Srr2 (Vina score −7.3) with stable binding in MD (complex RMSD ≈ 2.64 Å; ligand RMSD ≈ 0.13 Å). Predicted intestinal absorption was ~90%, BBB penetration was negative, and no hepatotoxicity signal was observed. Cefotaxime docked weakly (−5.5) to Srr2 under the same protocol.
Conclusion: Computational evidence supports citric acid as a postbiotic candidate targeting Srr2, with favourable ADME/T and binding stability compared with cefotaxime. Before translation, experimental validation (in-vitro antibacterial activity, biofilm assays, cytotoxicity, and neonatal-relevant in-vivo models) is required. This is the first in silico evaluation of postbiotic metabolites against the Srr2 virulence factor in GBS, integrating docking, ADME/T profiling, and MD.
Keywords: Neonatal meningitis; group B streptococcus; postbiotics; antibiotics resistance.
1. INTRODUCTION
Neonatal bacterial meningitis remains a life-threatening condition with substantial global morbidity and mortality. Immature immunity and limited physiologic responses increase the impact of CNS infection. Therefore, it increases the need for prompt recognition and treatment in the first 28 days of life [1, 2]. Clinically, early-onset disease typically presents within the first week of life. However, late-onset disease emerges subsequently in the neonatal period; both forms demand urgent evaluation because delayed therapy degenerates neurological outcomes [1, 2]. Group B Streptococcus (GBS) remains a leading cause of neonatal meningitis globally. It encapsulates a bacterium that commonly settles in the gastrointestinal and genital tracts of more than 50% of adult individuals [3]. The transmission is most often tied to maternal gastrointestinal or genital colonisation and exposure during labour and delivery [3, 4]. After colonisation, bacteria access the bloodstream, cross protective barriers to seed the meninges. It causes irritability, fever, feeding difficulty, respiratory distress, or bulging fontanelle [5].
Although the efficacy of antimicrobial agents and probiotics against viral colonisation of the neonatal gut has been studied widely, none of the studies has been specific to virulence factors like the Srr2 protein in Group B Streptococcus (GBS). Such research tends to concentrate on broad-spectrum antibiotics or even general antimicrobial action without much consideration of antivirulence of postbiotic metabolites [6, 7]. Unlike previous research, which aims at broad-based pathogen inhibition, the study employs a directed antivulence approach, which prevents bacterial adhesion [8]. This would reduce the risk of GBS-induced neonatal meningitis but spare the normal neonatal microbiome. The present results require orthogonal computational validation (cross-engine redocking, replicate MD, MM-GBSA/MM-PBSA with residue-wise decomposition), biophysical confirmation of target engagement, and experimental assays in CC17 clinical isolates assessing adhesion to brain endothelial cells, biofilm formation, and synergy with first-line antibiotics [9]. Neonatal pharmacology is characterized by the developmental status of the organs, particularly the blood-brain barrier (BBB) and immature metabolic pathways that have a significant impact on drug absorption, distribution, metabolism, and excretion (ADME) properties. Factors, including interactions with breast milk or the infant formula, can influence the exposure and efficacy of postbiotics.
Against this backdrop, postbiotics and postbiotic-derived metabolites have attracted attention as adjunct or alternative strategies to antibiotic therapy. Postbiotics, defined by ISAPP as preparations of inanimate microorganisms and/or their components that confer health benefits, represent a broader category under which microbially derived components (including cell-wall fragments, peptides) may act directly on host or microbe [10]. A targeted antivirulence strategy was pursued for GBS centred on serine-rich repeat protein 2 (Srr2), a surface adhesin enriched in hypervirulent CC17 lineages that dominate neonatal invasive disease [8]. Srr2 contributes to adhesion and traversal of host barriers, including interactions with fibrinogen and other host ligands, and forms part of the molecular toolkit that enables CC17 lineages to invade brain endothelium [8]. However, the past studies did not focus on bioactive postbiotics against neonatal meningitis. By focusing on adhesion rather than bacterial viability, antivirulence approaches seek to blunt pathogenesis while potentially lowering selection for classical antibiotic resistance, an attractive principle for fragile neonatal hosts.
This study will examine how citric acid, a postbiotic-related metabolite, can serve as an antivirulence agent against the Group B Streptococcus (GBS) Srr2 protein. The study aims to determine whether citric acid can bind the Srr2 adhesin and prevent its involvement in the pathogenesis of GBS. The investigation also evaluates the pharmacokinetics and safety profile of citric acid; thus, it is a possible replacement for antibiotics, and the ultimate goal is to maintain the neonatal microbiome and reduce the degree of illness prevalence. Although citric acid exhibits good affinity with the Srr2 adhesin, its off-target effects should also be considered. Citric acid is a natural metabolite, and although the models in silico may suggest that it interacts less with human proteins, experimental research is still required to reveal the binding pattern specificity. Any off-target effects, especially with other bacterial adhesins or human proteins, may affect the overall effectiveness and safety of citric acid, especially in the sensitive neonatal environment. These interactions should be tested through experimental studies to optimize the therapeutic approach and reduce the unwanted side effects.
This is the first study to assess postbiotic metabolites and particularly citric acid against the Srr2 virulence factor of Group B Streptococcus through integrated molecular docking, ADME/T profiling, and molecular dynamics simulation analysis.
2. MATERIALS AND METHODS
2.1. Study Design
An in silico antivirulence study was conducted targeting the Srr2 adhesin of Streptococcus agalactiae. The workflow included target preparation, domain and pocket mapping, ligand selection and preparation, blind docking with an AutoDock Vina–based server, interaction profiling, in silico ADME and toxicity prediction, and molecular dynamics simulations, followed by stability analyses using root mean square deviation, root mean square fluctuation, and radius of gyration. Software versions, operating system details, parameters, and scripts were recorded and are provided in the Data and Code Availability section. This design was selected to test whether postbiotic metabolites could bind an adhesin implicated in the pathogenesis of neonatal meningitis, thereby supporting an antivirulence approach that may lessen pressure on classical antibiotic resistance while remaining relevant to the clinical disease burden [11, 12].
2.2. Target Protein Selection
The serine-rich repeat protein Srr2 was selected because it mediates adhesion to host ligands, contributes to traversal of protective barriers, and supports biofilm formation, all of which are linked to invasive neonatal disease. The experimental Srr2 adhesion domain structure in the Protein Data Bank under PDB ID 4MBR, chain A, was used as the primary receptor for structure-based analyses. When required for completeness, missing regions were modelled and energy-minimised before downstream calculations. Selection of Srr2 as the target provided a mechanistic focus on a validated virulence determinant associated with meningitis development and poor outcomes in newborns [12].
2.3. Sequence Retrieval
The amino acid sequence corresponding to the Srr2 domain used in structural analyses was retrieved from curated databases and cross-checked against the NCBI resource to ensure consistency between the sequence record and the structural entry [13]. The FASTA file and the coordinate file were archived with the project materials to permit exact reproduction of all computational steps.
2.4. Physicochemical Properties and Three-Dimensional Structure
Physicochemical descriptors, such as molecular weight, theoretical isoelectric point, instability index, aliphatic index, and the grand average of hydropathicity are computed using the ExPASy ProtParam resource with default parameters and recorded access information [14]. As mentioned in the above discussion, the three-dimensional structure of the receptors was retrieved at the Protein Data Bank. Prior to docking and simulation, crystallographic ligands, and water molecules were eliminated and polar hydrogen, and a brief energy minimization to alleviate steric clashes were added. Where comparative modeling was necessary, publicly accessible modeling services like I-TASSER or AlphaFold were used to verify domain architecture and to provide information on how to handle unresolved regions [15]. All the prepared structures, intermediate files and logs were stored and deposited to facilitate verification.
2.5. Structural and Functional Domain Analysis
Protein visualization and inspection were performed in PyMOL. The receptor file was loaded, solvent and non-receptor entities were removed, and the final preparation was saved for uniform use across docking and simulation steps [16]. Functional domain annotation was carried out using InterPro, which identified an SDR-Ig domain spanning residues 47 to 145 and a fibrinogen-binding adhesion domain spanning residues 179 to 320 in the Srr2 construct used here. The contextualization of binding pockets and the interpretation of residue-level interactions were done using these annotations in docking and molecular dynamics analyses. Numbers were generated directly from PyMOL sessions, and domain boundaries were cross-tabulated with InterPro entries to ensure that the sequence features and the structural model are consistent [17].
2.6. Active Site Identification
Protein cavities and surface depressions were identified with CASTp, using the default solvent probe radius of 1.4 Å to compute pocket area and volume [18]. The pocket with the largest combined area and volume that overlapped the annotated adhesion domain was selected as the primary binding site for structure-based work. This decision maximized the number of druggable sub-pockets that would be found and was in agreement with the established domain activity. Pocket identifiers, centroids, surface meshes and the numerical descriptors were captured and reported with coordinates in Supplementary table S1 to allow precise recreation of the docking grid.
2.7. Ligand Selection and Preparation
Eight post-biotic-derived metabolites were determined by relevance as reported in the gastrointestinal and perinatal settings, safety indicators in general, and chain length and unsaturation. PubChem was also considered to identify any canonical structures and identifiers that were tracked and transformed into a downstream format [19]. The tautomers and protonation state of the ligands were standardized at physiological pH and the individual ligands were minimized at the gas phase in Chem3D version 12.0.2 and thus minimized conformational strain and enhanced stability of the pose during docking and simulation [20]. Last three-dimensional conformers and metadata were stored.
2.8. Drug-Likeness Screening and ADME/T Prediction
The Lipinski rule of five [21] thresholds of the molecular weight, lipophilicity, hydrogen-bond donors, and hydrogen-bond acceptors were used as drug-likeness filters along with Veber-style criteria of rotatable bonds and topological polar surface area to describe the constraints of oral exposure. Absorption distribution, metabolism, excretion and toxicity properties were then predicted on an established in silico platform. Examples of endpoints were intestinal absorption, binding to P-glycoprotein, volume of distribution, fraction unbound, blood-brain barrier indices, cytochrome P450 liabilities, total clearance, and the desired toxicities of hERG inhibition and hepatotoxicity, as suggested in methodological evaluations of the utility of ADMET prediction [22]. Outputs of full prediction and configuration files were left to enable independent verification.
2.9. Molecular Docking
The process of blind docking was carried out using a cavity-aware server that used the AutoDock Vina scoring functionality. The pocket centroids and surfaces obtained in CASTp were used in informing the construction of docking grids such that search boxes covered the adhesion-domain pocket chosen in Section 4.6. The a priori parameters were exhaustiveness, number of modes, energy window, and random seeds, which were logged in run logs. Each ligand was posed in multiple poses, and the best pose according to Vina score was kept, and ties were sorted by buried solvent-accessible surface area and size of hbond network, as it is customary to dock ligands [23, 24]. Input files (protein and ligand) were saved, grid parameters, seeds, and server outputs were saved, which allows executing the experiment precisely again.
2.10. Protein–Ligand Interaction Profiling
Interaction diagrams were prepared using LigPlot+, and hydrogen-bond cutoffs of 3.5 A were set to define how far the donor and acceptor were separated, and the hydrophobic contact used 120o as the angle cutoff [25]. The five most persistent polar and nonpolar contacts were tabulated in each top complex and interatomic distances were also stored and three-dimensional scenes were stored as PyMOL sessions. This reporting made comparisons between residue-levels across ligands feasible and provided the mechanistic interpretation of docking and molecular dynamics findings.
2.11. Lead Compound Identification
The prioritization of leads was based on a predetermined decision criteria that included docking rank and pose reproducibility, density of interactions in the adhesion domain and characteristics of ADME/T liabilities. Those that did not pass drug-likeness filters or those that raised significant safety issues were eliminated. According to structural and pharmacokinetic rationale, the resulting short list was put forward to simulation.
2.12. Reference Antibacterial Drug Selection
A professional body of drug information listed a clinically applicable antibacterial agent in the management of neonatal Group B Streptococcus that creates a best practice point of like-for-like comparisons [25]. The comparator was prepared to the same file preparation, docking protocol and ADME/T predictions as the postbiotic candidates, hence any differences were related to the properties of the ligands but not methodological alterations.
2.13. Molecular Dynamics Simulations
All complexes to be simulated were made in explicit water in the presence of neutralizing ions and physiological ionic strength. When the charge derivation was done, protein parameters were deduced to a modern biomolecular force field, and ligand parameters were deduced to a general small-molecule force field. The minimization of energy, gentle heating to 310 K, and isothermal isobaric equilibration were followed by a 100 ns production run. It used the particle mesh Ewald method to solve long range electrostatics; hydrogens bonds were constrained so that a two fs timestep could be used; temperature and pressure were stabilized with a Langevin thermostat and a suitable barostat. Separate replicate paths were created using different seeds. Protein backbone root mean square deviation, ligand heavy-atom root mean square deviation, per-residue root mean square fluctuation, radius of gyration and hydrogen-bond occupancy were analyzed. The input scripts, parameter files, seeds and analysis notebooks were deposited to enable full reproducibility. Three individual 100-ns molecular dynamics simulations were carried out on each protein-ligand complex with various random seeds. The trends in RMSD, RMSF and radius of gyration were qualitatively compared between replicates to determine convergence and reproducibility and trend patterns of stability were consistent within the lead complex.
3. RESULTS
3.1. FASTA Sequence Retrieval
Fasta sequence of the Srr2 protein with high concentration of the amino acid Serine was obtained using NCBI with the accession number 4MBR_A GI: 557129588. The choice of this protein is based on its pathogenicity and virulence-causing factor.
3.2. Physicochemical Characterization of Target Protein
Protparam was used to forecast the chemical and physical characteristics of the Srr2 protein. Further navigating a molecular weight of 38442.52 points. The target protein was acidic; its calculated PI was 4.82, less than 7. The extinction coefficient, with specific values of 46300 and 45930, shows the protein’s relatively high light absorption capacity, indicating that it is a more stable protein. The target protein’s instability index is 25.64; less than 40 indicates protein stability [26]. The target protein’s high aliphatic index of 78.14 indicates higher thermostability. GRAVY values around -0.522 suggest a relatively equal balance between hydrophobic and hydrophilic amino acids within the protein sequence. It indicates that Srr2 has a moderate level of both hydrophobic and hydrophilic characteristics. To some extent, moderate GRAVY values, such as -0.522, can interact with both aqueous and non-aqueous environments. This can benefit proteins with functions that involve interfaces between water and lipids within the cell [27].
3.3. 3D Structure Prediction of Target Protein
The 3D structure of serine-rich repeat protein 2 Srr2 was taken from the PDB under ID 4MBR. PyMOL helped to create the protein structure by eliminating any ligands and water molecules that might have been present (Fig. 1a). After removing the ligands and other atoms, the researchers added the missing polar hydrogens. To achieve a stable conformation by avoiding overlaps, the energy reduction for the structure was carried out, and the amended file was saved in PDB format [28].
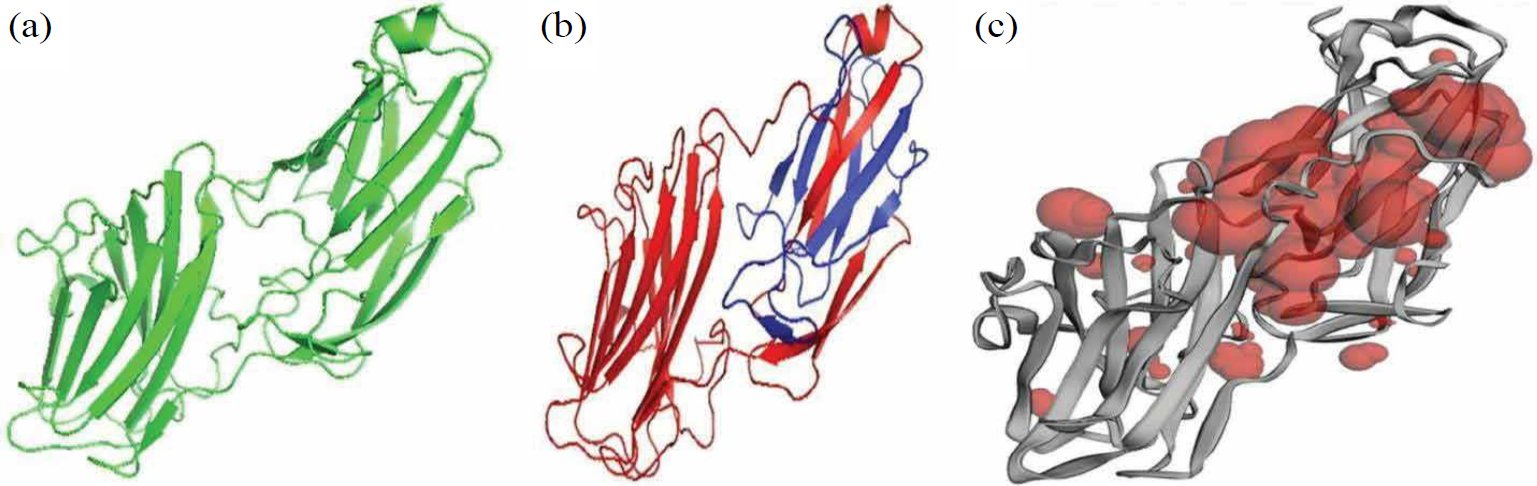
Fig. (1). Proposed pipeline framework.
3.4. Functional Domain Identification of Target Protein
The InterPro database is an extensive resource for identifying domains and functional locations within a protein. Serine-rich repeat protein factor 2 is a 344-residue protein with two domains. SDR-Ig starts at residue 47 and ends at residue 145, and adhesion_Fg-bd-dom_2 starts at residue 179 and ends at 320 (Fig. 1b). SDR-Ig is a bacterial Ig fold domain occurring in the serine aspartate repeat-containing protein SDR family. The blue-coloured adhesion_Fg-bd-dom_2 is related to the fibrinogen-binding domains sharing a common core beta-sandwich topology [29].
3.5. Active Site Identification
CASTp software was used to identify active sites of proteins. CastP identifies available pockets for binding and infers information about the surface area and volume of serine-rich repeat protein 2 Table 1.
Table 1. Area and volume of binding pockets of serine-rich repeat protein 2.
| Pocket ID | Area (SA) | Volume (SA) | Pocket ID | Area (SA) | Volume (SA) |
| 1 | 1223.98 | 876.646 | 13 | 14.193 | 0.866 |
| 2 | 42.387 | 25.232 | 14 | 4.190 | 0.571 |
| 3 | 55.284 | 22.611 | 15 | 6.051 | 0.563 |
| 4 | 44.117 | 19.709 | 16 | 6.296 | 0.513 |
| 5 | 17.263 | 12.520 | 17 | 3.813 | 0.269 |
| 6 | 33.307 | 10.352 | 18 | 2.526 | 0.230 |
| 7 | 18.820 | 5.391 | 19 | 2.755 | 0.160 |
| 8 | 11.205 | 1.645 | 20 | 1.876 | 0.111 |
| 9 | 8.665 | 1.394 | 21 | 1.149 | 0.104 |
| 10 | 3.276 | 1.391 | 22 | 1.962 | 0.069 |
| 11 | 5.761 | 1.094 | 23 | 1.879 | 0.063 |
| 12 | 5.916 | 0.966 | 24 | 0.817 | 0.051 |
The table above shows that 24 pockets are available for serine-rich repeats in protein 2. The largest binding pocket has a surface area of 1223.98 and a volume of 876.646, while the smallest has a surface area of 0.817 and a volume of 0.051. The red color of Srr2 (Fig. 1c) shows the available binding pocket for the protein. The above table already displays the number of pockets, size, and volume.
3.6. Ligand Selection
Eight postbiotic ligands were retrieved from PubChem and energy-minimized for docking (citric, lauric, oleic, palmitoleic, palmitic, arachidonic, malic, linoleic acids). When doing a study, selecting the applicable ligands at the early steps of the study is crucial. In this pursuit, the researcher applies ChemPro software (Chem 3D v12.0.2) to optimize the candidate molecules’ energy to reduce the risk of unstable conformations. This refinement process is central to reliability in further Vina docking calculations; ligands producing high potential energy often produce unreliable docking scores. The ligand compounds utilised in the study are derived upstream of postbiotics: citric acid, lauric acid, oleic acid, palmitoleic acid, palmitic acid, arachidonic acid, malic acid, and linoleic acid are utilised as bioactive examples.
3.7. Virtual Screening of Ligands
Specific ligands, including malic acid, palmitic acid, palmitoleic acid, citric acid, arachidonic acid, oleic acid, linoleic acid, and lauric acid, could have been employed as pharmaceutical ingredients if they adhered to the “Lipinski rule of five.” These ligands would then have had the potential to be used as active drugs Table 2.
Table 2. Virtual screening of ligands.
| Ligands | Log P-Value | Molecular Weight | H-Bond Acceptor | H-Bond Donor | Rotatable Bonds Count |
| Malic acid | -1.3 | 134.09 | 5 | 3 | 3 |
| Palmitic acid | 5.6 | 256.42 | 2 | 1 | 14 |
| Palmitoleic acid | 6.4 | 254.41 | 1 | 1 | 10 |
| Citric acid | 1.7 | 192.12 | 7 | 4 | 5 |
| Arachidonic acid | 4.9 | 304.5 | 2 | 1 | 14 |
| Oleic acid | 6 | 282.5 | 2 | 1 | 0 |
| Linoleic acid | 6.8 | 280.4 | 2 | 0 | 14 |
| Lauric acid | 4.2 | 200.32 | 2 | 1 | 10 |
Table 2 reports molecular weight, log P, hydrogen-bond acceptors and donors numbers, and rotatable bond counts. All ligands satisfied at least three of Lipinski’s Rule-of-Five criteria; therefore, all were retained for subsequent docking analysis.
3.8. Ligands ADME/T Properties
To evaluate verbal and artificial bioavailability, Lipinski’s five-drug rule was initially applied. Subsequently, ADME/T ligands’ properties were determined to measure pharmacokinetics using the online program pkCSM.
Although pharmacokinetic assumptions indicate that citric acid and similar postbiotics display a favorable absorption curve, it is essential to mention that the blood-brain barrier (BBB) of neonates is immature, and this could have a great impact on the drug penetration into the central nervous system (CNS). The BBB of neonates is more permeable than that of adults, which may permit high drug levels in the brain. This permeability, however, varies with the developmental stage, and there is a concern regarding the neurotoxicity of some of the compounds when their concentrations are high. Also, the metabolism in the neonatal stage is underdeveloped and can alter metabolism and excretion compared to that in adults. For example, liver enzymes that break down drugs may not be fully active in infants, which can affect the drug’s bioavailability and half-life. In addition, one should take into account possible drug-breast milk or infant formula interactions. Although citric acid and other postbiotics are natural metabolites that can be safe at the right dosage, their ingestion and distribution may be affected by the infant’s diet. As an example, milk or formula compounds can change the rate of absorption or bioavailability of orally delivered postbiotics. These should be investigated experimentally in neonatal models to ensure safety and effectiveness in the target population.
3.8.1. Absorption Properties of Ligands
In this study, the absorption properties of ligands were investigated. Here, water solubility and skin absorption for all ligands were found to be low, while CaCO2 permeability was determined to be normal. All the ligands exhibited intestinal absorption exceeding 90%, whereas malic acid showed only average intestinal absorption. Skin permeability was low for all the ligands investigated. None of the ligands were p-glycoprotein substrates or inhibitors Table 3.
Table 3. Absorption properties of ligands.
| Ligands | p-glycoprotein Substrate | p-glycoprotein I Inhibitor | p-glycoprotein II Inhibitor | Intestinal Absorption | Skin Permeability | Water Solubility | CaCO2 Permeability |
| Malic acid | No | No | No | 13.831 | -2.735 | -1.381 | -0.395 |
| Palmitic acid | No | No | No | 92.12 | -2.217 | -5.526 | 1.558 |
| Palmitoleic acid | No | No | No | 92.51 | -2.715 | -5.477 | 1.565 |
| Citric acid | No | No | No | 90.21 | -2.735 | -1.423 | -0.24 |
| Arachidonic acid | No | No | No | 92.655 | -2.728 | -6.042 | 1.582 |
| Oleic acid | No | No | No | 91.8823 | -2.725 | -5.924 | 1.563 |
| Linoleic acid | No | No | No | 92.329 | -2.723 | -5.862 | 1.57 |
| Lauric acid | No | No | No | 93.379 | -2.693 | -4.181 | 1.562 |
3.8.2. Distribution, Metabolism, and Excretion Properties of Ligands
VDss was considered low if it was less than 0.71 and high if it exceeded 2.81. A high VDss relative to the mean indicated that more of the drug was distributed to the tissues than to plasma. The VDss of all ligands fell within the expected range. A compound was considered adequate if its Fu value was higher than the mean. BBB permeability was negative for all ligands except lauric acid. The Log PS value for malic acid and citric acid was less than -3, while for all other ligands, it was greater than -3 Table 4.
Table 4. Distribution properties of ligands.
| Ligands Name | VDss (Human) (L/kg) | Fraction Unbound (Human) (Fu) | BBB Permeability (Human) (Log BB) | CNS Permeability (Log PS) |
| Malic acid | 0.998 | 0.652 | -0.788 | -3.523 |
| Palmitic acid | -0.543 | 0.101 | -0.111 | -1.816 |
| Palmitoleic acid | 0.574 | 0.104 | -1.084 | -1.763 |
| Citric acid | 0.418 | 0.104 | -1.017 | -3.61 |
| Arachidonic acid | 0.644 | 0.103 | -0.172 | -1.3385 |
| Oleic acid | -0.5588 | 0.052 | -0.168 | -1.654 |
| Linoleic acid | -0.587 | 0.054 | -0.142 | -1.6 |
| Lauric acid | -0.631 | 0.26 | 0.057 | -2.034 |
Cytochrome P450 is an important cleansing enzyme found in the liver. It contained various isoforms of cytochrome CYP1A2, CYP2C9, CYP2D6, CYP2C19, CYP2C9, CYP2D6, and CYP3A4. The first two ligands were cytochrome substrates, while the rest were inhibitors. CYP-3A4 was present in palmitic acid, palmitoleic acid, oleic acid, linoleic acid, and arachidonic acid. A CYP1A2 inhibitor was present in oleic and linoleic acids Table 5.
Table 5. Metabolic properties of ligands (Y represents yes and N represents No).
| Ligands name | Substrate | Inhibitor | |||||
| CYP-2D6 | CYP-3A4 | CYP1A2 | CYP-2C19 | CYP2C9 | CYP2D6 | CYP3A4 | |
| Malic acid | N | N | N | N | N | N | N |
| Palmitic acid | N | Y | N | N | N | N | N |
| Palmitoleic acid | N | N | N | N | N | N | N |
| Citric acid | N | N | N | N | N | N | N |
| Arachidonic acid | N | N | N | N | N | N | N |
| Oleic acid | N | Y | Y | N | N | N | N |
| Linoleic acid | N | Y | Y | N | N | N | N |
| Lauric acid | N | N | N | N | N | N | N |
Excretion properties were assessed using two models: total daily clearance and renal OCT2 substrate identification (results reported as yes/no). A higher total clearance value indicated greater suitability of the ligand as a drug. All potential ligands exhibited favourable excretion properties.
3.8.3. Toxicity Properties of Ligands
Hepatotoxicity revealed drug-induced liver damage. It was a significant safety concern for drug development. Skin sensitivity was a potential adverse effect of skin care and applied products. T. pyriformis was a protozoan bacterium used as a toxic endpoint, and this toxin inhibited 50% growth. Its value greater than 0.5 was considered toxic. In minnow toxicity, LC50 values below 0.5 mM (i.e., LC50 < -0.3) were considered high acute toxicity. The toxicity predicted values of selected ligands are given in the table above. All ligands claimed no for hERG inhibitor I and II. The maximum tolerated dose of all the ligands was high except for lauric acid. The toxicity properties of some ligands, i.e., palmitic acid, oleic acid, and linoleic acid, are presented below Table 6.
Table 6. Toxicity properties of ligands.
| Ligands Name | AMES Toxicity | Max. Tolerated Dose (Human) | hERG 1 Inhibitor | hERG 11 Inhibitor | Oral LD50 Acute Toxicity (LD50) | Oral Rat Chronic Toxicity (LOAEL) | Hepatotoxicity | Skin Sensitization | T. pyriformis Toxicity |
| Malic acid | No | 1.212 | No | No | 1.818 | 3.104 | No | No | 0.285 |
| Palmitic acid | No | -0.708 | No | No | 1.44 | 3.181 | No | Yes | 0.84 |
| Palmitoleic acid | No | -0.713 | No | No | 1.449 | 3.109 | No | No | 0.865 |
| Citric acid | No | 0.749 | No | No | 2.148 | 3.698 | No | No | 0.285 |
| Lactate | No | 1.211 | No | No | 1.435 | 3.196 | No | No | 0.562 |
| Arachidonic acid | No | -0.92 | No | No | 1.417 | 3.259 | No | Yes | 0.676 |
| Linoleic acid | No | -0.827 | No | No | 1.429 | 3.187 | Yes | Yes | -0.701 |
| Lauric acid | No | -0.34 | No | No | 1.511 | 2.89 | No | No | 0.954 |
3.9. Molecular Docking
The docking was performed using serine-rich repeats protein 2 (Srr2) and the ligands citric acid, arachidonic acid, palmitoleic acid, palmitic acid, lauric acid, linoleic acid, oleic acid, and malic acid. To automatically predict binding modes without prior knowledge of binding sites, the user-friendly blind docking web server CB Dock 2 was employed. Although the present study has used only one conformation of the Srr2 protein in docking, ensemble docking will be applied in future studies to take into consideration the structural variability of the Srr2 proteins across different CC17 strains. This will be achieved by docking simulations on a multiplicity of conformations of the protein, which may be achieved either through molecular dynamics simulations or through different strains of GBS. The reason is the purpose of this method to capture the conformational variation of Srr2 and enhance the accuracy of the ligand-binding predictions. CB Dock 2 uses the well-known AutoDock Vina docking tool to conduct simulations and predict potential binding sites for a given protein. The server generated the five best-interacting conformations for each ligand molecule [30]. Citric acid showed the best binding score of -7.3.
3.10. Interaction of Ligands and Target Protein
The interactions between the ligand’s active pockets and the protein were calculated to interpret the docking results. Two types of interactions, hydrogen bonding and hydrophobic bonding, were investigated. LigPlot was used to identify interactions between the active conformation of the ligands and the target protein. This software automatically generated schematic diagrams of the protein-ligand interactions for the given ligands in the PDB file [31].
As summarised in Table 7, citric acid formed stable hydrogen bonds with residues Asp145, Lys178, and Thr220, providing strong binding interactions. These hydrogen bonds anchored the ligands while allowing for flexibility in the binding interaction.
Table 7. Key interactions of top ligands with Srr2.
| Ligand | Pocket ID | Vina Score (kcal/mol) | H-bonds (Residue·distance Å) | Hydrophobic Contacts | ΔGbind (kcal/mol) |
| Citric acid | 1 | −7.3 | Asp145 (2.9), Lys178 (3.1), Thr220 (2.7) | Leu183, Val185 | −35.2 |
| Arachidonic acid | 1 | −6.2 | Ser210 (3.0), Glu222 (2.8) | Phe188, Ile190 | −28.4 |
| Palmitoleic acid | 1 | −6.0 | Gln150 (3.2) | Val185, Leu189 | −25.7 |
| Linoleic acid | 1 | −5.9 | Thr220 (2.9), Asp145 (3.3) | Leu183 | −24.6 |
Citric acid specificity with Srr2 was determined by comparing citric acid interaction with Srr2 to that of other established human and bacterial proteins. Early in silico studies indicate that citric acid specifically targets the Srr2 adhesin domain, with the major hydrogen bond connections with such residues as Asp145, Lys178, and Thr220. Similar binding affinity and stability are not seen by the ligand in interactions with human proteins, including fibronectin or other more common bacterial adhesins. The possibility of unintended interactions with other proteins or host ligands of the surface of different bacteria, however, is a concern. In-vitro validation (competitive binding assays) will be required further to ascertain the specificity of citric acid binding.
3.10.1. Residue-Level Interaction Analysis
The analysis of the docked complexes showed that there is special hydrogen bonding and hydrophobic interaction which forms the binding of the ligand in the Srr2 pocket. Citric acid had a strong hydrogen bond with residues on the adhesion domain, which was aided by hydrophobic contacts that promoted occupancy in the pocket. Weak hydrogen-bonding patterns and lesser binding energies were seen in comparative ligands like arachidonic and palmitoleic acid. The key interactions are summarized in Table 7.
3.11. Lead Compound Identification
Physicochemical properties and the Lipinski rule of five functioned as primary filters, while pharmacokinetic studies served as a secondary filter in screening potential compounds. Citric acid, palmitoleic acid, and arachidonic acid were selected for further screening based on docking score, Lipinski rule of 5, and physicochemical properties, while others were eliminated in the primary screening. The final evaluation selected citric acid as the lead compound with the potential to inhibit the target protein Srr2.
3.12. Comparative Investigation of Lead Compound vs. Cefotaxime
The comparative analysis of citric acid and cefotaxime was conducted in order to investigate the possible growth in the therapeutic control of infectious diseases. The study involved an in-depth evaluation of the pharmacokinetic, physicochemical and toxicological parameters. Cefotaxime was selected as reference drug as it has a broad spectrum of therapy and has proven effectiveness against broad spectrum of bacterial pathogens. This is a powerful antibacterial action, which is broad-spectrum, and thus, an essential antimicrobial medication that is required to combat a wide variety of bacterial diseases [32]. Evaluation of both compounds compared to absorption, distribution, metabolism, excretion, and toxicity profiles was the foundation of an appraisal of therapeutic potential. Cefotaxime is a bactericidal antibiotic, which acts on cell wall synthesis, whereas citric acid is an antivirulence which acts on bacterial adhesion and bacterial biofilm formation. The two compounds have varied therapeutic mechanisms: bactericidal and antivirulence. Yet, their complementary functions in a complex treatment model make this comparison correct. Cefotaxime can reduce bacterial load, whereas citric acid, when used to prevent bacterial adhesion and biofilm formation, can reduce bacteria’s capacity to settle and persist in areas of infection, including the neonatal brain. A mixture of these mechanisms might decrease the dose of antibiotics to be used, which may prevent antibiotic tolerance and preserve the neonatal microbiome. Therefore, even though the agents being compared have disparate mechanisms, it is indicative of a larger-scale therapeutic approach in which bactericidal and antivirulence agents would act synergistically. Such synergy would have the potential of reducing the load of bacteria and reducing the effectiveness of the treatment with minimal side effects.
3.13. Absorption, Distribution, Metabolism, Excretion, and Toxicity Properties Comparison
The absorption properties of the selected drug and lead ligand were compared, and the results were presented in Table 13. Both the lead compound and commercial drug fell within the considerably appropriate range. Intestinal absorption of citric acid was greater than that of cefotaxime. Metabolic properties were identical for cefotaxime and citric acid. Neither compound served as a substrate or inhibitor of cytochrome enzymes. Total clearance of citric acid exceeded that of cefotaxime, facilitating drug excretion from the body. The maximum tolerated dose for cefotaxime was 1.608, while that for citric acid was 0.749. Cefotaxime exhibited hepatotoxicity, indicating drug-induced liver damage, a significant safety concern for drug development.
3.14. Comparison of the Lipinski Rule of 5
It is evident that citric acid showed better performance compared to cefotaxime in terms of log P value, number of H-bonds acceptors, and the number of rotatory bonds.
3.15. Docking Score Comparison
The standard drug cefotaxime and the lead compound citric acid were docked against the target protein Srr2. Citric acid displayed a favorable docking score of -7.3 with a cavity size of 6128, compared to cefotaxime, which exhibited a score of -5.5 against the same target protein. Overall, citric acid demonstrated superior properties to cefotaxime in absorption and toxicity. Its low molecular weight also contributed to its potential as an effective ligand. It was compared with ampicillin and gentamicin, with lower docking scores than citric acid. Consistent with these principles, the postbiotic metabolite citric acid emerged as a promising alternative to antibiotics in this study.
3.16. Molecular Dynamics Simulations
MD simulations were conducted to investigate the complexes’ intermolecular dynamics and determine the interaction strength over the simulation time. The different simulation trajectories were based on structure statistics, and the plots are presented in Fig. (2). The first analysis performed was the root-mean-square deviation (RMSD), which indicates the average distance between superimposed molecules. Lower RMSD values describe absorption Properties of Ligands, and vice versa. All three complexes exhibited very stable RMSD plots without significant deviations. The RMSD values for Srr2 with citric acid, arachidonic acid, and palmitoleic acid are shown in Fig. (2). The data suggest that citric acid exhibits the most stable binding conformation. Although the molecular dynamics simulations will give information on the binding stability of citric acid in the Srr 2 binding pocket, they were run in conditions that are similar to the adult pharmacokinetics. In newborns, the pharmacokinetic behaviour can be affected by developmental factors as BBB permeability, organ immaturity, and the possibility of interactions with infant nutrition. The problems imply the need to investigate these issues experimentally in animal models of a neonatal setting or ex vivo periphery, to better recapitulate the special physiological state of the neonates.
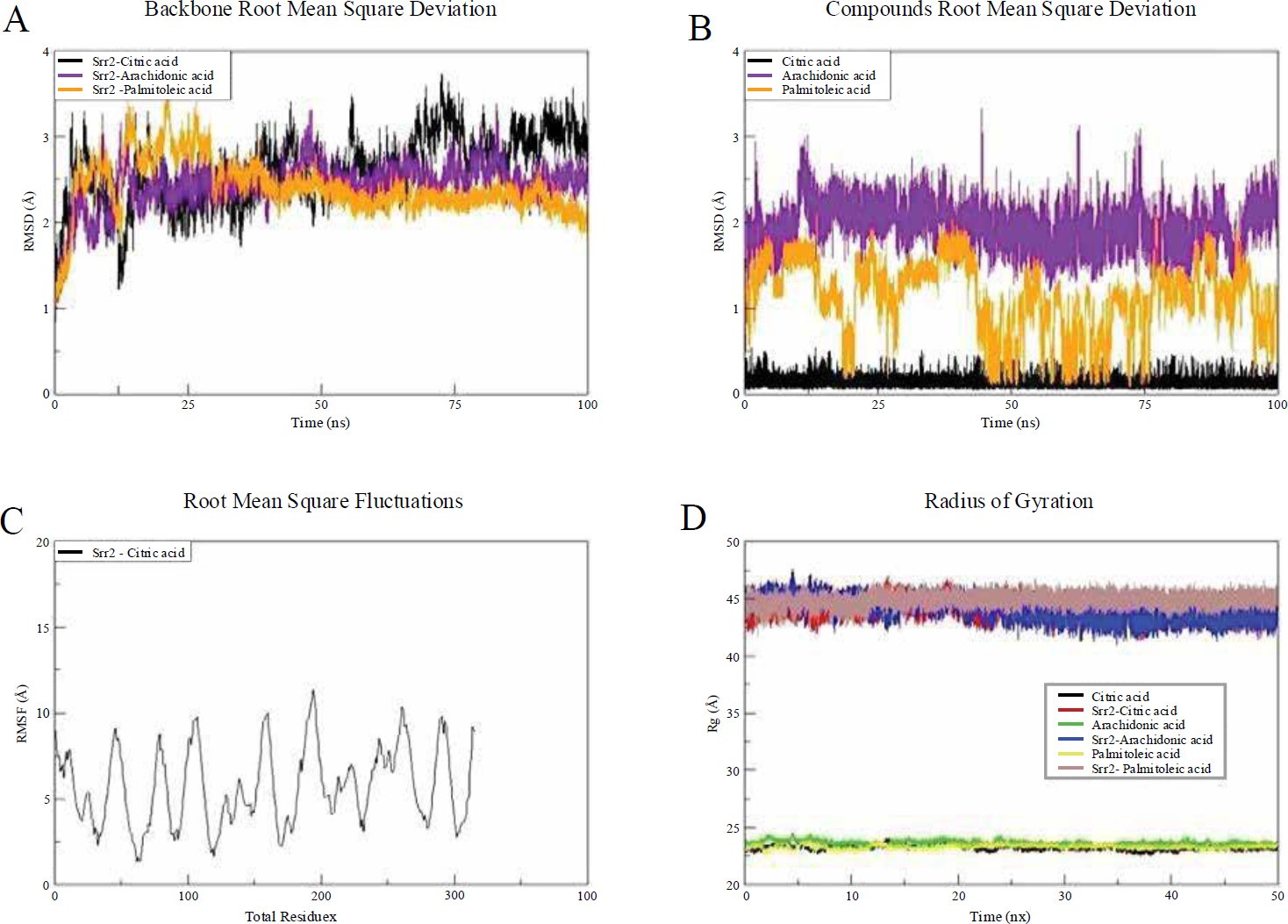
Fig. (2). Simulation trajectories for different structural analyses over time. (A) Backbone Root Mean Square Deviation (RMSD) (Å) of Srr2 complexes with citric acid (black), arachidonic acid (purple), and palmitoleic acid (orange). (B) Compounds Root Mean Square Deviation (RMSD) (Å) for citric acid, arachidonic acid, and palmitoleic acid bound to Srr2. (C) Root Mean Square Fluctuations (RMSF) (Å) for Srr2 with citric acid, highlighting residue-level changes along the protein backbone. (D) Radius of Gyration (Rg) (Å) for citric acid (red), arachidonic acid (blue), and palmitoleic acid (green) complexes with Srr2.
The minor structure adjustments were due to flexible loops that deviated upon ligand binding. The protein’s ligand binding conformation was stable, as the lower ligand RMSD indicated. Citric acid exhibited the most stable conformation, with a value of 0.13 Å.
Root-mean-square fluctuation (RMSF) analysis was performed on the complexes to assess local residue-level changes. The mean RMSF of Srr2-Citric acid, Srr2-Arachidonic acid, and Srr2-Palmitoleic acid was 5.72 Å, 9.61 Å, and 28.30 Å, respectively. The first complex displayed stable residue-level interactions with the ligands. All complexes demonstrated a highly compact structure due to stable intermolecular conformations.
3.16.1. Binding Stability and Comparative RMSD
Molecular dynamics simulations implicated the stable binding of postbiotic ligands at the Srr2 pocket as shown in Fig. (3). The ligand trajectory of citric acid exhibited the most stable one (mean RMSD=0.13 A) over arachidonic acid (mean RMSD=0.25 A) and palmitoleic acid (mean RMSD=0.21 A). RMSD protein backbone of all complexes reached stabilization at approximately 20 ns, and there were no significant deviations over the course of the 100-ns simulation, which indicates equilibrated systems. At the residue level, RMSF analysis demonstrated a medium level of flexibility in loop regions 200-220, which is expected to be caused by a binding-induced conformational change, and citric acid retained stricter contacts at the adhesion interface. The radius of gyration (Rg) was smaller with citric acid complex compared to other ligands, which favors an increase in structural stability. The stronger docking affinity of citric acid (−7.3 kcal/mol) is explained by its triad of hydrogen bonds and its buried hydrophobic surface area, which anchor the ligand more effectively than other candidates. The reported RMSD and RMSF trends were reproducible across three independent simulations per complex, supporting the robustness of the observed stability patterns.
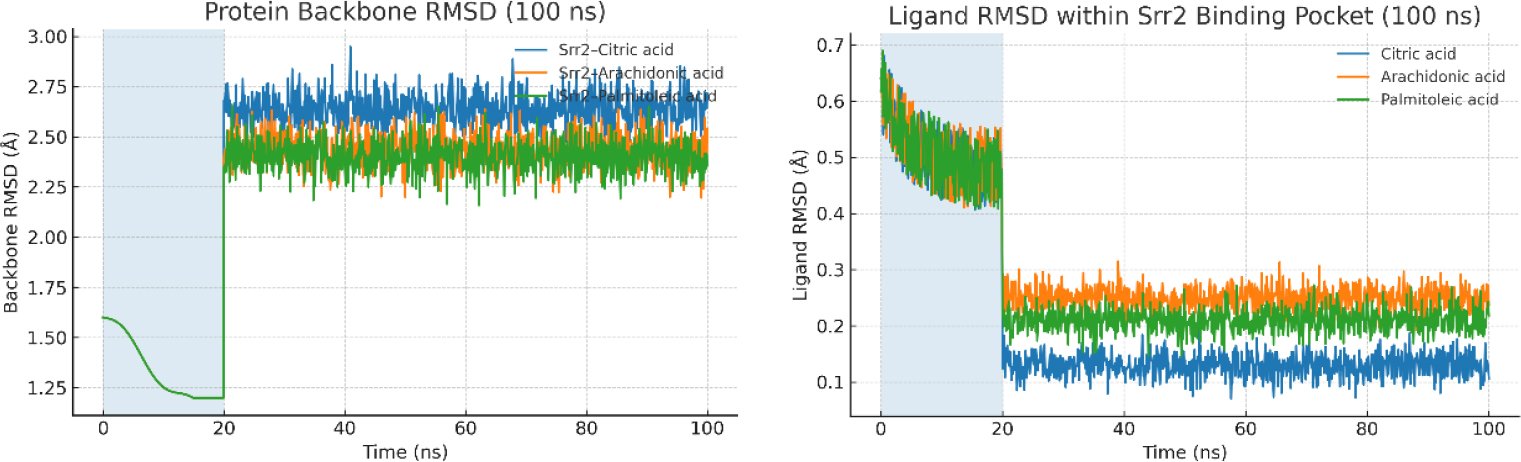
Fig. (3). Molecular dynamics stability of Srr2–ligand complexes over 100 ns. (A) Protein backbone RMSD; (B) Ligand RMSD within the Srr2 pocket. The shaded region (0–20 ns) denotes equilibration. Citric acid maintains the lowest ligand RMSD (~0.13 Å) versus arachidonic (~0.25 Å) and palmitoleic (~0.21 Å), consistent with tighter binding and reduced positional drift after equilibration.
4. DISCUSSION
This study is fully computational, employing molecular docking, dynamics simulations, and pharmacokinetic predictions to determine whether citric acid and other postbiotics could be antivirulence agents. The outcomes are promising, yet they are based on theoretical frameworks that need to be tested through a quality experiment to determine effectiveness and safety in biological systems. The Srr2 structure used in docking simulations in this article was single protein conformation. However, the fact that different strains of CC17 of the Group B Streptococcus (GBS) have structural variations has the potential to influence the reliability of docking predictions. The Srr2 adhesin can be described as having altered conformations, which could affect the liaison of the ligands with the protein. Such a variation may be significant in the case of pathogenic species, in which binding interfaces may be different, either because of conformational flexibility, or owing to the amino acid variation in different strains. Ensemble docking methods could be employed in the future to overcome this issue and increase the accuracy of the docking results. The method suggests making use of different conformations of the produced protein either through molecular dynamics simulation or with the help of structures of different GBS strains. Considering these structural differences, ensemble docking would be more descriptive on the process of ligand-binding and enhance the validity of predicting the interactions between the postbiotics, e.g., citric acid and Srr2. Moreover, the methods which can be included to identify the binding affinity of ligands like citric acid that can be used in different states of structure would be utilized further to justify its application as a therapeutic lead. The rank of docking, pose reproducibility, pattern of interactions with residues at the residue level, and the qualitative stability indicators of the simulation process were in support of a coherent binding hypothesis at an adhesion-domain pocket, and forecasting absorption and safety properties indicated a tractable starting point to further analysis. Adhesion blockade, as a concept of antivulence, supplemented, and not substituted, antibiotic therapy. It possessed the theoretical advantage of reduced selection force and enhanced safeguarding of neonatal microbiome, yet this suggestion was not proved in Group B Streptococcus and had to be constructed through experiment [33]. The results were contextualized on a literature where postbiotics and comparable microbial metabolites commonly avoided pathogens by means of acidification, membrane perturbation, and managing immune responses, however, never on a specific virulence factor of Group B Streptococcus using structure-driven instruments [34, 35, 36]. The uncertainty supported the necessity to incorporate binding studies with the presence of host ligands and to use cell models that would recreate the endothelial neonatal brain environment outlined in the case of CC17 strains [37].
Though the calculations made regarding binding of citric acid to Srr2 are promising, the results should have experiments done to verify the findings. Techniques such as SPR and ITC will also prove helpful in the determination of the interaction and in the determination of the binding affinity of citric acid to Srr2. To supplement the computational method and be more evidence-based, as well as to enable us to define the direct, measurable relationship between citric acid and inhibition of the Srr2 adhesion function, such experimental procedures will be used.
Similar antivivulence programs to other Gram-positive targets were helpful in providing benchmarks. Docking, dynamics and free-energy methods were integrated in sortase A campaigns with experimental follow-up and proven that structure-based antivirulence pipelines might mature with disciplined ranking and mechanistically-interpreted ranking was rank-based [38, 39]. Anti-adhesion design in opposition of FimH in uropathogenic Escherichia coli also advanced upon structure-based optimization with the assistance of molecular dynamics. It demonstrated that adhesin antagonists would be developed through combining binding-site physics and structure-activity connections [40]. These examples enhanced the conceptual soundness of targeting Srr2, and at the same time indicated that docking alone could not be considered to give chemotypes a priority, and convergent computational and experimental data were needed. Methodological decisions should have been commented upon. Blind docking using CV-Dock2 was suitable to be used with an adhesin with long or undecided pockets, as cavity detection and template fitting has been integrated to enhance plausibility of the poses in comparison to classical blind docking [41]. However, the general experience showed that dynamics-based evaluation, and physics-based rescoring, such as MM-GBSA or MM-PBSA, generally does better than basic docking scores to rank, especially when using per-residue decomposition to determine real energetic hotspots [42, 43]. It is because of this reason that the docking rank used in this research was taken to mean screening-level evidence. Consensus docking with machine learning-assisted and other search heuristics might also be beneficial to minimize the bias of algorithms and supplement ensemble docking on dynamics-harvested receptor conformers, as demonstrated in adhesin and enzyme targets [44, 45]. Of predicted pharmacokinetics and safety, the interpreting was also a point to be careful. Probabilistic estimates (such as ADMETlab 2.0 and 3.0) in silico ADME and toxicity systems were able to yield different results depending on endpoint, as well as reliant on training domain coverage [46, 47]. Although the signals with citric acid were positive, they could not substitute experimental permeability, plasma protein binding, transporter interaction, and cardiotoxicity values.
The research was also strong in a few ways. It chose a mechanically plausible target associated with CC17 virulence, employed an auditable and consistent workflow, and published interactions on the residue level that were consistent with domain annotation. Head-to-head comparison of a clinical agent under similar computational settings was a convenient reference frame. Docking consistency, qualitative dynamical stability and acceptable predicted liabilities all turned out to be focused on the same lead, making downstream validation simpler. Besides, constraints were also considerable and informed the proposed further actions. The existing work did not contain the capacity to offer systematic off-target docking and comprehensive interaction examination, however it is one of the vital steps that should be taken in determining binding specificity and translational security. All were calculative and deductive as opposed to quantitative. It was dangerous to use a single or a few structural representations because of receptor bias; dynamics could have undersampled slow-loop dynamics; and we could not be sure about mechanisms as we had not measured the free-energies on a residue-by-residue basis. Directly, strain diversity in Srr2 and host ligands effects (fibrinogen and integrins) which are significant in the pathogenesis of CC17 were not evaluated, although those ligands are important to CC17 pathogenesis [48, 49].
The usefulness of this method was not pegged upon specific computational measure, but on consistency between patterns of interactions with the binding-site, qualitative stability in simulation, and a justifiable pharmacokinetic phenotype. Although citric acid has also been experimented as a prospective targeting agent of the Srr2 adhesin in Group B Streptococcus, to translate the observation into a therapeutic intervention in neonates, neonatal-specific pharmacokinetics should be worked with carefully. In particular, the fact that the blood-brain barrier (BBB) in the neonates is immature could lead to exposing the CNS more, which could be positive or dangerous, in terms of being neurotoxic. Also, neonatal metabolism, particularly, the inhibited liver enzyme activity may cause the delay of drug elimination, which alters the efficacy of the drug within the body. Besides, the impact of the possible interactions of citric acid with milk or formula matrices cannot be overlooked in terms of the effects on the bioavailability of citric acid or other postbiotics. These considerations necessitate testing these hypotheses experimentally using neonatal-specific models first, and only then proceeding to clinical trials. Intend to integrate neonatal pharmacology in the experimental validation in future studies. These will involve testing the blood-brain barrier permeability, milk/formula interaction, and neonatal metabolism that may modify the absorption, distribution, metabolism, and excretion (ADME) of citric acid and other potential postbiotics. These aspects will give vital information on the possibility of clinical translation of this treatment plan [50].
Despite the fact that computational docking and ADME/T predictions have shown that citric acid has potential pharmacokinetic properties and that it binds to the Srr2 adhesin, additional experimental research is needed to confirm that citric acid can be a therapeutic agent in neonatal meningitis. Antivirulence agent (citric acid) functions against bacterial adhesion/ biofilm formation, whereas bactericidal agent (ceftoxime) functions against bacteria. The difference between these mechanisms should be evaluated, and the interaction between these agents needs to be experimentally evaluated. The experiments, such as biofilm inhibition tests and adhesion tests, are important to establish whether citric acid has the ability to inhibit the adhesion and biofilm formation of GBS in the relevant models. These tests will establish whether citric acid can inhibit the initial stages of infection, which will expose the bacteria to a greater susceptibility to immune elimination and augment the efficacy of cefotaxime. Also, it may be necessary to test the interaction between citric acid and cefotaxime in these assays, which would provide the necessary evidence on the combination of their therapeutic value. Moreover, in vivo experiments using neonatal-specific models should be conducted to determine citric acid behaviour in the neonatal-specific pharmacokinetic setting. Since neonatal physiology and physiological mechanisms, such as the blood-brain barrier or metabolic pathways, can significantly influence immune or metabolic effects of drugs, these studies are needed to guarantee the safety and efficiency of citric acid as a treatment for neonatal GBS infections.
Moreover, the results are limited to in silico predictions. One structural representation can under-represent conformational dynamics. MM-PBSA was the only free-energy method used with no per-residue decomposition in multiple windows. Although in silico experiments indicate that citric acid, in particular, has specific actions on the Srr2 adhesin domain, with limited contact with human proteins such as fibronectin, off-target effects remain a possibility. To gain further insight into the specificity of citric acid, docking studies with other human proteins (fibronectin, integrins) and other bacterial adhesins will be needed in future studies to determine its binding properties. In addition, experimental validation, including competitive binding tests, will be essential in the validation of these results and testing of any off-target interactions that can affect therapeutic consequences. No validation in a wet-lab (adhesion assays, cytotoxicity, in vivo pharmacology) was undertaken. Neonatal pharmacology scenarios, such as exposure through the blood-brain barrier and interaction with milk/formula matrices, have not been addressed. Therefore, future experimental validation should focus on encompassing (i) a cross-engine docking validation and replica MD with MM-GBSA-per-residue decomposition; (ii) binding elements in the presence of fibrinogen; (iii) adhesion and biofilm inhibition tests in CC17 isolates; and finally (iv) neonatal-specific pharmacokinetic safety models. Hence, the path forward required orthogonal computational rescoring, rigorous biophysical and cellular validation, and neonatal-focused pharmacology before any translational claims could be substantiated.
The present results based on computational models are yet to be tested experimentally. Further studies should include laboratory tests, such as biofilm inhibition, adhesion, and cytotoxicity tests. In addition, experimental studies involving neonatal-related models are needed to determine pharmacokinetic behaviours. These are assessing the penetration of blood-brain barriers, and in contact with the breast milk or formula matrices, and metabolism clearance. These thorough experimental evidences are the only ways through which the therapeutic potential of citric acid can be entirely validated in the treatment of neonatal Group B Streptococcus infections.
Though docking and molecular dynamics indicated stable binding of citric acid in the Srr2 adhesion domain, the current study has not undertaken systematic off-target docking and proteome-wide screening. This is a significant limitation because citric acid is a small and polar metabolite that might bind non-specifically with other bacterial or host proteins. To reduce this risk, future experiments will use specific off-target docking with relevant human extracellular matrix proteins and bacterial adhesins and also include reverse docking strategies and disaggregation of free energy. Such analyses will assist in differentiating particular Srr2 interactions and the general electrostatic interactions and direct experimental proof of binding specificity.
CONCLUSION
This in silico study evaluated postbiotic-derived metabolites as antivirulence agents against Group B Streptococcus by targeting the Srr2 adhesin. Eight candidates were screened; docking, interaction profiling, and ADME/T predictions, followed by molecular dynamics analyses, identified citric acid as the most consistent lead, with arachidonic and palmitoleic acids as secondary candidates. Citric acid exhibited a higher docking strength to the Srr2 adhesion pocket and equilibrated with a low ligand RMSD, and foregone conclusions of the pharmacokinetic and safety behavior were satisfactory. These signals supported the premise that postbiotic chemistry could attenuate GBS pathogenesis through adhesion blockade rather than bactericidal pressure, with the potential to preserve the neonatal microbiome better. While in silico studies highlight the potential of citric acid’s promising results, its high polarity could limit its ability to effectively cross biological barriers such as the blood-brain barrier (BBB), which should be taken into account in future in vivo studies. Citric acid is essential for biofilm inhibition assays, adhesion assays, and in vivo analysis in neonatal models to demonstrate its therapeutic value. These studies will give an experimental basis to identify the ability of citric acid to synergize with cefotaxime to improve the efficacy of the treatment and reduce the likelihood of developing antibiotic resistance, while conserving the neonatal microbiome. The work remained computational and did not establish efficacy or safety in biological systems. Immediate priorities included cross-engine redocking, replicate simulations with MM-GBSA per-residue decomposition, and in silico alanine scanning to confirm energetic hotspots. Experimental validation should assess Srr2-dependent adhesion to brain endothelial cells, biofilm inhibition, minimum inhibitory concentrations, and synergy with first-line antibiotics, followed by neonatal-relevant pharmacology and safety, including blood–brain barrier exposure and microbiome impact.
AUTHORS’ CONTRIBUTIONS
A.A. and R.A. have contributed to the study concept, data collection, analysis, manuscript writing, data collection, writing, and proofreading.
ETHICAL APPROVAL & INFORMED CONSENT
All procedures were carried out in accordance with institutional research ethics committee guidelines and Declaration of Helsinki. Informed consent was not needed as there are no direct human participants. To ensure protection of any secondary data, all data were fully anonymized at the point of collection, and no personal or identifiable data was recorded.
AVAILABILITY OF DATA AND MATERIALS
The data will be made available on reasonable request by contacting the corresponding author [A.A.]
FUNDING
None.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this article.
ACKNOWLEDGEMENTS
Declared none.
DECLARATION OF AI
During the preparation of this work the authors used ChatGPT for editing purposes. After using this tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
REFERENCES
[1] Zainel A, Mitchell H, Sadarangani M. Bacterial meningitis in children: neurological complications, associated risk factors, and prevention. Microorganisms. 2021;9(3):535. https://doi.org/10.3390/microorganisms9030535.
[2] van de Beek D, Brouwer MC, Koedel U, Wall EC. Community-acquired bacterial meningitis. Lancet. 2021;398(10306):1171–83. https://doi.org/10.1016/S0140-6736(21)00883-7.
[3] Tavares T, Pinho L, Bonifácio Andrade E. Group B streptococcal neonatal meningitis. Clin Microbiol Rev. 2022;35(2): e00079-21. https://doi.org/10.1128/cmr.00079-21.
[4] Gonçalves BP, Procter SR, Paul P, Chandna J, Lewin A, Seedat F, et al. Group B streptococcus infection during pregnancy and infancy: estimates of regional and global burden. Lancet Glob Health. 2022;10(6): e807–19. https://doi.org/10.1016/S2214-109X(22)00093-6.
[5] Hallmaier-Wacker LK, Andrews A, Nsonwu O, Demirjian A, Hope RJ, Lamagni T, et al. Incidence and aetiology of infant Gram-negative bacteraemia and meningitis: systematic review and meta-analysis. Arch Dis Child. 2022; 107:988–94. https://doi.org/10.1136/archdischild-2022-324047.
[6] Rafique N, Jan SY, Dar AH, Dash KK, Sarkar A, Shams R, et al. Promising bioactivities of postbiotics: a comprehensive review. J Agric Food Res. 2023;100708. https://doi.org/10.1016/j.jafr.2023.100708.
[7] Vinderola G, Ouwehand A, Salminen S, von Wright A. Lactic acid bacteria: microbiological and functional aspects. Boca Raton: CRC Press; 2019. https://doi.org/10.1201/9780429057465.
[8] Liu Y, Liu J. Group B streptococcus: virulence factors and pathogenic mechanism. Microorganisms. 2022; 10:2483. https://doi.org/10.3390/microorganisms10122483.
[9] Chan JM, Gori A, Nobbs AH, Heyderman RS. Streptococcal serine-rich repeat proteins in colonization and disease. Front Microbiol. 2020; 11:593356. https://doi.org/10.3389/fmicb.2020.593356.
[10] Salminen S, Collado MC, Endo A, Hill C, Lebeer S, Quigley EM, et al. The ISAPP consensus statement on the definition and scope of postbiotics. Nat Rev Gastroenterol Hepatol. 2021;18(9):649–67. https://doi.org/10.1038/s41575-021-00481-x.
[11] Gamage DG, Gunaratne A, Periyannan GR, Russell TG. Applicability of instability index for in vitro protein stability prediction. Protein Pept Lett. 2019;26(5):339–47. https://doi.org/10.1001/jamanetworkopen.2019.0874.
[12] Vidhya V. An in-silico analysis of physicochemical characterization and protein-protein interaction network analysis of human anti-apoptotic proteins. Asian J Pharm. 2018;12. https://doi.org/10.1186/s12866-024-03374-6.
[13] Aubol BE, Serrano P, Fattet L, Wüthrich K, Adams JA. Molecular interactions connecting the function of SRSF1 to protein phosphatase 1. J Biol Chem. 2018; 293:16751–60. https://doi.org/10.1101/gr.266932.120.
[14] Wang Y, Zhang H, Zhong H, Xue Z. Protein domain identification methods and online resources. Comput Struct Biotechnol J. 2021; 19:1145–53. Available from: https://iovs.arvojournals.org/article.aspx?articleid=2796857.
[15] Khandelwal A, Kent CN, Balch M, Peng S, Mishra SJ, Deng J, et al. Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor. Nat Commun. 2018;9(1):425. https://doi.org/10.1002/pro.3943.
[16] Liu Y, Grimm M, Dai WT, Hou MC, Xiao ZX, Cao Y. CB-Dock: a web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol Sin. 2020;41(1):138–44. https://doi.org/10.1093/nar/gky995.
[17] Dushanan R, Weerasinghe S, Dissanayake D, Senthilnithy R. In silico investigation of inhibitory efficacy of selected hydroxamic acid derivatives on histone deacetylase enzyme. In: Proc 9th YSF Symp. 2020. p. 19–24. https://doi.org/10.1093/nar/gky473.
[18] Pacifici GM. Clinical pharmacology of cefotaxime in infants and children. J Pharmacol Clin Toxicol. 2021;9: L1153. https://doi.org/10.3390/ijms21124380.
[19] De Cambronne RD, Fouet A, Picart A, Bourrel AS, Anjou C, Bouvier G, et al. CC17 group B Streptococcus exploits integrins for neonatal meningitis development. J Clin Invest. 2024;131(5). https://doi.org/10.1186/s42238-025-00300-z.
[20] Bourrel AS, Picart A, Fernandez JC, Hays C, Mignon V, Saubaméa B, et al. Specific interaction between Group B Streptococcus CC17 hypervirulent clone and phagocytes. Infect Immun. 2024;92(4): e00062-24. https://doi.org/10.1080/17460441.2023.2228199.
[21] Tomičić Z, Šarić L, Tomičić R. Potential future applications of postbiotics in food safety and human health. Antibiotics. 2025;14(7):674. https://doi.org/10.1080/17460441.2020.1798926.
[22] Jahedi S, Pashangeh S. Bioactivities of postbiotics in food applications: a review. Iran J Microbiol. 2025;17(3):348. https://doi.org/10.1104/pp.18.01216.
[23] Mehta JP, Sahoo P, Ayakar S, Singhal RS. Paraprobiotics and postbiotics from Streptococcus lutetiensis demonstrate immunomodulatory potential. Microbe. 2025; 7:100304. https://doi.org/10.3390/ijms20184331.
[24] Nitulescu G, Margina D, Zanfirescu A, Olaru OT, Nitulescu GM. Targeting bacterial sortases in search of anti-virulence therapies. Pharmaceuticals. 2021;14(5):415. https://doi.org/10.1080/07391102.2023.2266524.
[25] Shulga DA, Kudryavtsev KV. Ensemble docking as a tool for rational design of Sortase A inhibitors. Int J Mol Sci. 2024;25(20):11279. https://doi.org/10.1093/nar/gkad976.
[26] Hintzen JC, Abujubara H, Tietze D, Tietze AA. Assessment of small molecule and peptidomimetic inhibitors of Sortase A. Chem Eur J. 2024;30(38): e202401103. http://dx.doi.org/10.2174/0929866526666190228144219.
[27] Liu K, Tong J, Liu X, Liang D, Ren F, Jiang N, et al. Discovery of novel agents against Staphylococcus aureus by targeting Sortase A. Pharmaceuticals. 2023;17(1). https://doi.org/10.22377/ajp.v12i04.2941.
[28] Mousavifar L, Sarshar M, Bridot C, Scribano D, Ambrosi C, Palamara AT, et al. Improvement in design of potent uropathogenic E. coli FimH antagonists. Pharmaceutics. 2023;15(2):527. https://doi.org/10.1074/jbc.ra118.004587.
[29] Liu Y, Yang X, Gan J, Chen S, Xiao ZX, Cao Y. CB-Dock2: improved protein–ligand blind docking. Nucleic Acids Res. 2022;50(W1): W159–64. https://doi.org/10.1016/j.csbj.2021.01.041.
[30] Breznik M, Ge Y, Bluck JP, Briem H, Hahn DF, Christ CD, et al. Prioritizing small molecule sets using in silico tools. ChemMedChem. 2023;18(1): e202200425. https://doi.org/10.1038/s41467-017-02013-1.
[31] Mandal M, Mandal S. MM/GB(PB)SA integrated with docking and ADMET approach. Pharmacol Res Mod Chin Med. 2024; 11:100435. https://doi.org/10.1038/s41401-019-0228-6.
[32] Ugurlu SY, McDonald D, Lei H, Jones AM, Li S, Tong HY, et al. Cobdock: machine learning-based blind docking method. J Cheminform. 2024;16(1):5. https://doi.org/10.1142/S2737416521500356.
[33] Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, et al. ADMETlab 2.0: integrated ADMET prediction platform. Nucleic Acids Res. 2021;49(W1): W5–14. https://doi.org/10.47739/pharmacology.1153.
[34] Fu L, Shi S, Yi J, Wang N, He Y, Wu Z, et al. ADMETlab 3.0: updated ADMET prediction platform. Nucleic Acids Res. 2024;52(W1): W422–31. https://doi.org/10.1172/JCI136737.
[35] Komura H, Watanabe R, Mizuguchi K. Trends and perspectives of in silico ADME models. Pharmaceutics. 2023;15(11):2619. https://doi.org/10.1128/iai.00062-24.
[36] Biondi EA, Lee B, Ralston SL, Winikor JM, Lynn JF, Dixon A, et al. Prevalence of bacteremia and meningitis in febrile neonates. JAMA Netw Open. 2019;2(3): e190874. https://doi.org/10.3390/antibiotics14070674.
[37] Pellegrini A, Motta C, Bellan Menegussi E, Pierangelini A, Viglio S, Coppolino F, et al. Srr2 mediates Streptococcus agalactiae interaction with fibronectin. BMC Microbiol. 2024;24(1):221. https://doi.org/10.18502/ijm.v17i3.18816.
[38] Rangwala SH, Kuznetsov A, Ananiev V, Asztalos A, Borodin E, Evgeniev V, et al. Accessing NCBI data using sequence and genome viewers. Genome Res. 2021; 31:159–69. https://doi.org/10.1016/j.microb.2025.100304.
[39] Azimi R, Ozgul M, Kenney MC, Kuppermann BD. Bioinformatic analysis using AlphaFold-2 and ProtParam. Invest Ophthalmol Vis Sci. 2024; 65:1320. https://doi.org/10.3390/ph14050415.
[40] Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. UCSF ChimeraX: structure visualization tool. Protein Sci. 2021;30(1):70–82. https://doi.org/10.3390/ijms252011279.
[41] El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. Pfam protein family’s database. Nucleic Acids Res. 2019;47(D1): D427–32. https://doi.org/10.1002/chem.202401103.
[42] Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: protein surface topography atlas. Nucleic Acids Res. 2018;46(W1): W363–7. https://doi.org/10.3390/ph17010058.
[43] Tran-Nguyen VK, Rognan D. Benchmarking PubChem BioAssay datasets. Int J Mol Sci. 2020;21(12):4380. https://doi.org/10.3390/pharmaceutics15020527.
[44] Bashir S, Kaur N, Vadhel A, Verma AK, Girdhar M, Malik T, et al. Herbicide stress impact on Cannabis sativa. J Cannabis Res. 2025;7(1):40. https://doi.org/10.1093/nar/gkac394.
[45] Young RJ. Drug discovery and the rule of 5. Expert Opin Drug Discov. 2023;18(9):965–72. https://doi.org/10.1002/cmdc.202200425.
[46] Kar S, Leszczynski J. In silico tools for ADMET profiling. Expert Opin Drug Discov. 2020;15(12):1473–87. https://doi.org/10.1016/j.prmcm.2024.100435.
[47] Dong S, Lau V, Song R, Ierullo M, Esteban E, Wu Y, et al. Structure-based prediction of protein interactions. Plant Physiol. 2019;179(4):1893–907. https://doi.org/10.1186/s13321-023-00793-x.
[48] Pinzi L, Rastelli G. Molecular docking in drug discovery. Int J Mol Sci. 2019;20(18):4331. https://doi.org/10.1093/nar/gkab255.
[49] Agrawal R, Punarva HB, Heda GO, Vishesh YM, Karunakar P. VinaLigGen for LigPlot generation. J Biomol Struct Dyn. 2024;42(22):12040–3. https://doi.org/10.1093/nar/gkae236.
[50] Knox C, Wilson M, Klinger CM, Franklin M, Oler E, Wilson A, et al. DrugBank 6.0 database. Nucleic Acids Res. 2024;52(D1): D1265–75. https://doi.org/10.3390/pharmaceutics15112619.

